
Gene: MYO7A
Year Identified: 1995
Each research project listed below will include a graphic of the research continuum. The gold box indicates where this project falls on the continuum, illustrating its progress towards reaching people living with Usher syndrome, from "Bench to Bedside."
Click here to learn more about the different stages in the research continuum.
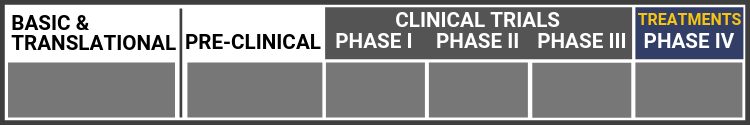
Gene Therapy for USH1B


Shannon Boye, Ph.D., and Atsena Therapeutics:
University of Florida & Founder, Director of Atsena Therapeutics
Developing a Dual AAV vector-mediated therapy for Myosin VIIa Usher Syndrome
Dr. Shannon Boye and the team at Atsena are making rapid advancements in evaluating a dual AAV vector-based gene therapy to prevent blindness from MYO7A-associated Usher Syndrome (USH1B). USH1B is caused by mutations in the MYO7A gene. USH1B patients are born profoundly deaf and begin to progressively lose vision in their first decade. The therapeutic gene to treat USH1B (MYO7A) is too large to fit inside a single AAV vector. To address this barrier, Atsena Therapeutics developed dual AAV vectors capable of delivering this large genetic payload, marking a potentially major advance in preventing blindness in patients affected by this devastating multisensory disorder.
Natural History Study in Subjects With Usher Syndrome Type 1B
The natural history study is being conducted to understand the progression of the disease in patients with USH1B as measured by a number of vision-related assessments. Disease progression will be evaluated as change over time in these measures, and associations between endpoints will be examined. They are recruiting patients in Naples, Italy, in Madrid, Spain, and Rotterdam, Netherlands.
Locations and Principal Investigators:
Francesca Simonelli
Naples, Italy
Eye Clinic of the University of Campania Luigi Vanvitelli
Email: Francesca.SIMONELLI@unicampania.it
Ingeborgh van den Born
Rotterdam, Netherlands
Stichting Oogziekenhuis Rotterdam
Email: born@oogziekenhuis.nl
Carmen Ayuso
Madrid, Spain
Instituto de Investigacion Sanitaria de la Fundacion Jimenez Diaz
Email: cayuso@fjd.es
Clinicaltrials.gov listing: Natural History Study in Subjects With Usher Syndrome (USH1B)
DUAL AAV VECTOR FOR GENE THERAPY OF USH1B RETINITIS PIGMENTOSA ("USHTHER")

Alberto Auricchio, MD:
TIGEM Naples, Italy
A consortium of researchers around Alberto Aurrichio has worked on dual AAV vectors for non-clinical studies and a clinical trial. Currently the safety and expression of dual AAV vectors is being assessed in primates. Additionally, a phase 1/2 clinical trial to investigate the safety and efficacy of dual AAV-MYO7A vectors in humans has been designed.
The objective of the study USHTher is to test the safety and efficacy of an innovative gene therapy approach that uses dual AAV in retinas of USH1B patients. This study is the first time the dual AAV vector has been used in humans. Twelve USH1B patients will be given the dual AAV vectors for the study. The hope is that this study will create the basis for a cure for USH1B retinitis pigmentosa (RP). The use of dual AAV vectors could also hopefully be applied to other conditions that require large genes.
UshTher Clinical Trial Fact Sheet
Dr. Alberto Aurricchio's Lab Page
Create a Nonhuman Primate Model of Usher Syndrome

Martha Neuringer, Ph.D.:
Oregon National Primate Research Center and Casey Eye Institute
Foundation Fighting Blindness Individual Investigator Research Award Recipient
Dr. Neuringer and her colleagues are using the gene-editing technique CRISPR/Cas9 to develop a large animal model of Usher type 1B, which is caused by mutations in the gene MYO7A. She believes that these animals will exhibit vision loss and will therefore be useful for testing potential Usher 1B therapies.
Gene Editing of the Usher 1B gene
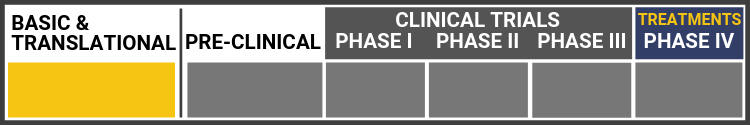

David Williams, Ph.D.:
UCLA Stein Eye Institute/Ophthalmology
Foundation Fighting Blindness Individual Investigator Research Award Recipient
Dr. Williams is using an innovative gene-editing technique called CRISPR/Cas9 for correcting a mutation in the gene MYO7A, which causes Usher syndrome type 1B. MYO7A is a large gene, making it difficult to replace with conventional (viral) gene replacement therapy.
Dr. David Williams' Lab Page
Link to Relevant Publication
Characterization of existing and newly developed models of Usher Syndrome

Jacque Duncan, MD:
University California, San Francisco
Experts in genetics and cell biology will create new models using zebrafish, 13-lined ground squirrels and retinal pigment epithelial cells from iPSC to study how the genes and proteins associated with Usher syndrome contribute to photoreceptor structure and survival.
Dr. Jacque Duncan's Lab Page
Generation, establishment and phenotypical characterization of porcine models for human Usher syndrome - USH1 pig models


Kerstin Nagel-Wolfrum, Ph.D. and Uwe Wolfrum, Ph.D.:
Kerstin and Uwe Wolfrum have developed a USH1B pig model. The researchers currently analyze a naturally occurring USH1B/MYO7A pig model bearing a biallelic nonsense mutation (p.Q181*) in the MYO7A gene. This project is in close collaboration with Michael Wendt, Hanover, and Doris Höltig, Berlin. They will make use of this model to define the mechanisms underlying the visual dysfunction in USH1B and to provide a large animal for preclinical evaluation of therapies for USH1B patients. Kerstin and Uwe Wolfrum are also working on a USH1C pig model.
Study of UshStat in Patients With Retinitis Pigmentosa Associated With Usher Syndrome Type 1B
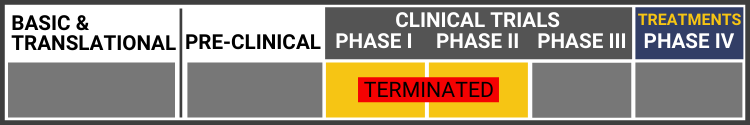
(TERMINATED: Study stopped not for safety reasons. Due to review of clinical development plans and priorities, Sponsor decided to stop development of the product.)
Clinical trials to evaluate the safety and the tolerability of ascending doses of subretinal injections of SAR421869 (UshStat) in USH1B patients are taking place in Paris, France at the Hopital Nationale des Quinze-Vingt and in Portland, Oregon at the Casey Eye Institute. It is gene replacement therapy where a correct copy of the MYO7A gene is injected into the retina through a lentiviral vector, which has the capacity to carry a large gene like MYO7A. However, since December 2018 clinical trials have been on hold in Phase 1/2 after Sanofi removed it from their pipeline. In the February 2019, press release they have announced they are seeking an out-licensing partner and/or co-sponsor.
There is a second trial that is following the long-term effects of those who have participated in the terminated SAR421869 trial.
Clinicaltrials.gov listing: Study of SAR421869 in Patients With Retinitis Pigmentosa Associated With Usher Syndrome Type 1B
Clinicaltrials.gov listing: A Study to Determine the Long-Term Safety, Tolerability and Biological Activity of UshStat® in Patients With Usher Syndrome Type 1B
USH1B-Related Science News
AAVantgarde is working on a new gene therapy and has launched a clinical trial for people living with Usher syndrome type 1B.
Researchers studied 80 people with Usher syndrome type 1B (USH1B) to learn how vision changes over time.
In late 2024, AAVantgarde shared exciting announcements on the first participant dosage, Orphan Drug Designation, and early safety data in the Phase 1/2 LUCE-1 clinical trial for vision loss caused by Usher syndrome type 1B.
To develop successful treatments, scientists must understand disease etiology and its corresponding biomarkers, which are biological molecules.
Italy’s Center for Rare Ocular Diseases at the University of Campania designed and conducted a longitudinal natural history study of European patients with confirmed Usher syndrome diagnosis and biallelic MYO7A variants.
Researchers wanted to learn more about how hearing loss occurs in individuals with changes in the MYO7A gene. They studied a large group of people in Japan who had hearing loss.
Usher syndrome (USH) is classified into three major clinical subtypes (types 1–3), with type 1 being the most severe.
This study used a special type of 3D imaging to look at tiny parts of inner ear cells in zebrafish.
Martha Neuringer, Ph.D., leads the research team at OHSU, Oregon Health & Science University, that confirmed the first-ever nonhuman primate model of Usher syndrome.
Dr. Neuringer's lab created a monkey with the MYO7A mutation that causes Usher Type 1B. For the first time, an animal model demonstrates all three phenotypes of USH1B: deafness, impaired balance and retinal degeneration.
This is significant because primates are the closest genetic cousins to humans, and having this animal model allows scientists to better understand Usher syndrome and test potential treatments.
MYO7A is a large gene that encodes myosin VIIA, a protein that helps maintain stereocilia in the inner ear and the retinal pigment epithelium in the retina.
Researchers used CRISPR/Cas9 technology to disrupt the MYO7A gene in monkeys to create a nonhuman primate model for Usher syndrome type 1B (USH1B).
Usher syndrome is the most common cause of deafness associated with visual loss of a genetic origin. The purpose of this paper is to report very severe phenotypic features of type 1B Usher syndrome in a Saudi family affected by a positive homozygous splice site mutation in MYO7A gene. This mutation manifested with advanced retinal degeneration at a young age.
What this means for Usher syndrome: Individuals with this particular mutation may experience more severe symptoms than other Usher 1B patients.
In this USH Talk, Dr. Shannon Boye summarizes efforts to develop a dual AAV vector-based gene therapy for Myosin7a Usher syndrome (USH1B). The drawbacks of USH1B mouse models and a rationale for testing these vectors in a more clinically relevant species are discussed.
Jennifer Phillips, Ph.D." on defining “Failure”: Disclosing when things don’t work and understanding WHY is a really important, though often overlooked realm of research. Here are a couple of USH1 research stories from today’s presentations that illustrate that point.
This review, published by Williams et al discusses possible gene therapy approaches for the prevention of retinal degeneration in Usher syndrome.
João Carlos Ribeiro, Bárbara Oliveiros, Paulo Pereira, Natália António, Thomas Hummel, António Paiva & Eduardo D. Silva
Study aimed at identifying and characterizing putative differences in olfactory capacity between patients with USH and controls, as well as among the subtypes of USH.
Hidekane Yoshimura, Maiko Miyagawa, Kozo Kumakawa, Shin-ya Nishio, and Shin-ichi Usami.
This first report describing the frequency (1.3–2.2%) of USH1 among non-syndromic deaf children highlights the importance of comprehensive genetic testing for early disease diagnosis.
Gene therapy is still a relatively new development, and so far, the only USH target being delivered via viral vector in clinical trials is MYO7A (USH1B). There are a few different reasons for this, but all boil down to a numbers game.
Three patients have been treated so far with no serious adverse events after six months. They have been allowed to proceed to delivering a larger dose to the next group of patients.
The bulk of the presentations I took in today were reports from clinicians treating Usher patients. I don’t get to interact with clinicians on a regular basis, so it is hugely instructive for me to get their perspective on diagnosis and monitoring of the progressive retinal degeneration seen in Usher syndrome
Today was an 11-hour maelstrom* of really good science. Of all the great research stories I heard, there are several that will likely be of interest to our readers:
At last year’s ARVO conference there was a presentation reporting successful animal testing for a gene therapy product called “UshStat”*. While this work has not yet appeared in a peer-reviewed publication the ARVO abstract can be found here. The poster presentation at the meeting described the use of a non-pathogenic viral vector to deliver a normal copy of the gene affected in Usher Type 1B (MYO7A) into the retina.
In preparing to write this blog post, I planned to leap right into the meat of the study, because I know that’s what you all are most keen to hear about. However, once I got into it I realized that I’d be doing our readers a disservice by cutting to the juicy center without context.
The US Food and Drug Administration (FDA) has approved Oxford BioMedica's Investigational New Drug (IND) application for the Phase I/IIa clinical development of UshStat to treat Usher syndrome type 1B. Oxford Biometica will enroll 18 patients with Usher type 1b at the Casey Eye Institute in Portland, Oregon. The study will be lead by Dr. Richard Weleber.
This 2010 review dives into the genetics of pathological mechanisms of Usher syndrome.
What this means for Usher syndrome: Research has come a long way since 2010!







