
Prelude to a Gene Therapy Report: Everything you ever wanted to know about Myosin VIIA
April 5, 2012
by Jennifer Phillips, Ph.D.
One of the many nice things about living in Oregon is the referred prestige of sharing a State with the Casey Eye Institute at Oregon Health Sciences University. Casey is the premier facility in the state for visual problems of all kinds and the clinicians there serve the majority of Usher patients in Oregon. Beyond the excellent clinical care, the breadth and quality of the research in various eye health areas conducted there are impressive indeed, and some of the promising Usher research that I’ve blogged about previously has originated from the professionals affiliated with Casey. Now, a new clinical trial, auspiciously the first human clinical trial to test a therapy targeting vision loss in Usher syndrome patients, is underway there.
In preparing to write this blog post, I planned to leap right into the meat of the study, because I know that’s what you all are most keen to hear about. However, once I got into it I realized that I’d be doing our readers a disservice by cutting to the juicy center without context. I’ve already told you the punch line (in bold, above) and given you a link to the press release, so if that’s enough information for you, feel free to stop reading and know that I will most certainly keep you updated on the results of this trial as soon as the data are available. But maybe a few of you—especially those who have a personal interest in USH1B in particular—might want to dig a little deeper into what this therapy is designed to do specifically, and HOW it might actually achieve that. With a little background, your understanding and appreciation of how we arrived at the point of being able to test such a therapy, the punch line might take on additional significance.
To best serve whatever number of our readers fit that description, therefore, I found myself faced with two choices: plunge ahead to produce a single post with such density that it would run the risk of developing its own gravitational pull, or break it up into a couple of more digestible posts. Not wanting to meddle with the laws of physics, I naturally gravitated (ha!) toward the second option. So, for the remainder of this post, I will tell you more about the affected gene in USH1B, what the molecule is thought to do for visual function under normal circumstances, and what, specifically, we think goes wrong in the retinas of USH1B patients. In the second part, I’ll describe the preclinical experiments that led to the current clinical trial and try to set up some expectations about what might come out of this Phase I/Phase IIa trial.
The gene that is affected in USH1B patients encodes a protein called Myosin VIIA. Anyone who’s had biology or anatomy in the past might recall that the prefix “myo” usually indicates an association with muscle, and indeed, the most widely known myosins are involved in muscle contractions. Myosins are a part of a pretty big protein family found in many species from single-celled organisms on up to complex critters like us. In general terms, myosins are referred to as ‘motor proteins’, i.e. they move stuff. In muscles, myosins drag muscle fibers around; in other cell types, myosins are responsible for a whole host of other molecular movements. So let’s look at Myosin VIIA (which I’ll hereafter abbreviate as MYO7A) in particular—in what cell types is it found? What might it be moving in there? And, importantly, how would failure to be at those locations moving those things result in Usher syndrome?
Remember this post? Like many of the Usher proteins, MYO7A localizes to the region of the connecting cilium, the structure that connects the part of the cell where proteins are produced to the part where many of them are required to function. Our best hypothesis right now is that the Usher proteins are required to load and move cargo—specifically molecules required for receiving and responding to light. What happens when these proteins don’t get to their proper locations? Well, functionally, the ‘receiver’ part of the photoreceptor, called the outer segment, won’t have the right equipment on board to do its job. Moreover, even though molecules aren’t being transported effectively, they are presumably still being made, and, absent a functional conveyance system, are likely piling up in the production part of the cell known as the inner segment. Having a buildup of protein in the wrong place can also cause problems for the cell.
Most everything we know about what Myo7a is thought to be doing in photoreceptors comes from research in mice bearing mutations in this gene. This type of mouse is also known as shaker1, so called for the pronounced balance defects that cause it to move unsteadily and shake its head. The Myo7a gene in shaker1 mice contains mistakes in the coding sequence for an important functional domain of the protein. From these mice, we’ve learned that Myo7a protein localizes to a couple of key cell types in the retina: photoreceptors and Retinal Pigmented Epithelial (RPE) cells. Like several of the other Usher mouse models, the shaker1 mice don’t experience retinal degeneration, despite having severe hearing and balance problems typical of Usher type 1. However, looking closely at the retinas of shaker1 mice reveals that things aren’t exactly 100% ok in there.
Let’s look at the data from photoreceptors first:
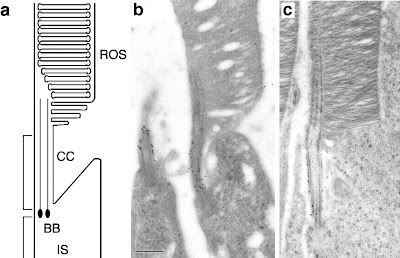
Panel A is a schematic of the region of the photoreceptor we’re looking at. From bottom to top, “IS” stands for Inner Segment, BB (basal body) and CC (connecting cilium) are components of the structure that conveys molecules from the IS to the outer segment. In this case, the depicted cells are rod photoreceptors, thus the top part of the cell is labeled “ROS” for Rod Outer Segment—the place where visual pigments gather in droves to await activation by photons.
Panel B is actually showing you two (rod) photoreceptors, but you can only see the outer segment in the one on the right. The CC, however, is present in both of them, and along that sinewy-looking vertical structure you can see a lot of little black dots. Those dots are Myo7a protein, and their abundant (relative to the sparse number of black dots visible in other parts of the cell) presence at the CC suggests a functional role in that region of the cell
Panel C is a (rod) photoreceptor from a shaker1 (Myo7a) mutant mouse. As you can hopefully see, there are black dots (Myo7a protein) around the connecting cilium—BUT, those black dots are labeling mutant protein. The mutation obviously didn’t affect the mechanism for bringing Myo7a to that region of the cell, but will its ability to function there be compromised? The next figure might help answer this question:
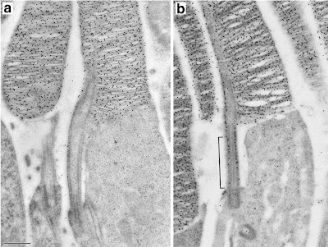
In panel A is a normal mouse rod, but this time the black dots aren’t Myo7a. In this series the black dots are labeling the protein rhodopsin, a visual pigment molecule found in abundance in the outer segment. The thing to notice in this panel is not so much where the black dots are as much as where they aren’t. Specifically, the connecting cilium (long skinny stalk-like thing in the middle) is pretty free of black dots. The mechanism by which opsins ascend the cilium is quite fast, so it is not surprising that the snapshot in time represented by this image didn’t catch very many rhodopsin molecules in the act of being transported.
Compare this to panel B. While there is lots of labeled rhodopsin up in the outer segment where it’s supposed to be, there’s also quite a lot hanging out in the region of the connecting cilium. This suggests that the opsin molecules aren’t being effectively transported, and from this we can conclude that Myo7a probably plays some role in the ciliary transport of opsin (and maybe other things, too).
As mentioned above, mouse mutants of Myo7a have also indicated that this protein is active in RPE cells. Before we delve into what it might be doing there, though, let’s briefly touch on the RPE itself. Where is it and what does it do? The Retinal Pigmented Epithelium is a layer of cells that lies just next to the photoreceptors. RPE cells contain a lot of black blobby looking things called melanosomes—basically, pigment filled sacs. The pigmentation in these cells helps them absorb light as it passes through the eye. RPE cells are also essential for the health and well-being of photoreceptors (indeed, a number of retinal degenerative diseases can result from problems in the RPE cells), and part of this function is achieved through the very close contact these cells establish with photoreceptor outer segments:

As depicted in Figure 3 above, the RPE cells are sort of like jellyfish, wrapping their tentacles around the photoreceptor outer segments. Instead of ‘tentacles”, we eye experts prefer the term ‘apical processes’. In a normal mouse RPE, These apical processes also contain melanosomes, as seen in panel A of Figure 4,below. This figure shows isolated (i.e. no longer associated with photoreceptor cells) RPE cells, so all the tissue that you see here corresponds to the blue jellyfish in Figure 3. The bracket on the right indicates the region containing the apical processes (aka tentacles). The important thing to note here is the relative abundance of melanosomes in this region—they look like little black cigars:
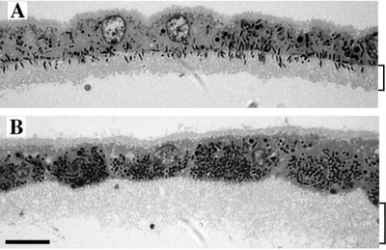
Panel B of the above image shows the same type of isolated RPE, with the region of apical processes again indicated by the bracket. This RPE is taken from a Myo7a mutant mouse, and what you can hopefully see without looking too hard is that the AP region doesn’t contain any melanosomes. All the little black cigars are staying up in the RPE cell bodies, again, indicating a problem with transport.
Despite these clear defects and beautifully detailed images, however, no retinal disease phenotype has been observed in shaker1 mice. Even though these mice clearly have some transport issues in not one but two retinal cell types, the photoreceptors don’t degenerate and the mice don’t lose their vision. At least that’s the way things stood until late last year.
A paper published in August 2011 (from some of the same researchers involved in the new gene therapy, as it happens) reports that when the shaker1 mutation is present in a genetic background that makes the animals more sensitive to light (notably not the same genetic background in which the above experiments were conducted), a specific defect in photoreceptors is observed. This defect is, again, related to transport—specifically, the light-dependent movement of a protein found in rod photoreceptors. Moreover, this more light-sensitive strain of mouse, when also mutant for Myo7a, experiences retinal degeneration that accelerates when the animals are exposed to brighter light levels.
From all these experiments, we can conclude that defects in Myo7a in mice lead to protein trafficking defects and, in certain genetic backgrounds, result in progressive retinal degeneration. We can further extrapolate from this that similar defects in moving proteins to the right places in retinal cells might be the cause of the retinal degeneration experienced by USH1B patients. One final important conclusion: In the light-sensitive genetic background, the shaker1 (Myo7a) mouse mutant can now serve as a model for the retinal degeneration seen in human Usher 1B patients.
In the follow-up post, I’ll describe the gene therapy experiments conducted with this animal model and how they paved the way for the clinical trials now underway at the Casey Eye Institute.
References:
Liu, X., Udovichenko, I.P., Brown, S.D.M., Steel, K.P., and Williams, D.S. (1999) Myosin VIIa Participates in Opsin Transport through the photoreceptor cilium. J Neuroscience 19(15):6267-6274.
Gibbs, D., Azarian, S.M., Lillo,C., Kitamoto, J., Klomp, A.E., Steel, K.P., Libby, R.T., and Williams, D.S. (2004) Role of Myosin VIIa and Rab27a in the motility and localization of RPE melanosomes. J Cell Science 117: 6473-6483.
Peng, Y-W., Zallocchi, M., Wang W-M., Delimont, D and Cosgrove, D. (2011) Moderate light-induced degeneration of rod photoreceptors with delayed transducin translocation in shaker1 mice. IOVS 52 (9): 6421-6427.







