
Dispatches from ARVO 2014, Closing Remarks: Designing gene therapies for rare diseases
May 16, 2014
by Jennifer Phillips, Ph.D.
Gene therapy is still a relatively new development, and so far, the only USH target being delivered via viral vector in clinical trials is MYO7A (USH1B). There are a few different reasons for this, but all boil down to a numbers game. MYO7A was likely chosen as the target for this therapy because it is a common type of USH and stands to help the most number of people. The patented gene replacement therapy being tested, UshStat, is a commercially produced pharmaceutical product. Research and development of such therapies are expensive undertakings, and there has to be reasonable certainty that a sufficient number of people will benefit (and, in some form, pay for) from a given therapy before company resources are devoted to moving it forward. In the standard, FDA-approved chain of development, there also have to be a sufficient number of people to test the therapy on in clinical trials. There are enough USH1B patients in the world to participate in several parallel, ongoing clinical trials of UshStat. It’s basic supply and demand law at work. And, as you’d expect, people with rare diseases are left out of the loop here—not just rarer forms of USH, but a variety of other afflictions that, as debilitating and lethal as they might be, just don’t affect enough people to justify putting industry R & D dollars into a gene-specific cure.
All that said, the number of people affected isn’t the only consideration. As many of you know, USH1B is not the most common type of USH overall. It is, however, the most common of all the USH genes that will actually fit into a viral vector. The most common type of all is USH2A, which is unfortunately far too large to work with a viral delivery system. Thus, the challenges we have, as people who have a personal or professional stake in helping USH patients, include:
- Finding ways to make gene- or disease-specific therapies available to everyone, regardless of the rarity of their disease type
- Developing therapies that are compatible with gene sizes that exceed the maximum capacity of the current viral delivery systems.
The past several years have seen amazing developments in the area of stem-cell-based research for retinal diseases. When I first began attending the ARVO meeting nearly a decade ago, the technology to maintain retinal cells in culture didn’t exist. The information needed to maintain differentiated retinal cells was difficult to come by, but a few years ago, the secret formula had been discovered—a long, complex cascade of gene expression was required to compel cells to take on a retinal identity, and survive in culture beyond that point. This enormous breakthrough was coupled with the equally important knowledge of how to compel functioning adult cells (skin fibroblasts, specifically) to forget everything they knew about how to be skin cells and revert to an undifferentiated stem cell-like state. Once undifferentiated, these induced pluripotent stem cells could then be compelled to differentiate as a new cell type. At this point, the obstacles that had limited stem cell research over the past couple of decades had been cleared, ushering in a new era of possibilities and potential new therapies for patients with degenerative diseases. In the ensuing years, more and more research labs have been able to harness this powerful cell culture model and, using adult skin biopsies, generate personalized stem cell lines or differentiated tissues in culture for use in disease research.
One of the premier research efforts with respect to the potential impact on Usher patients is taking place at the University of Iowa. The University of Iowa Department of Ophthalmology and Visual Sciences has been a mainstay of USH diagnosis and treatment for a number of years. In the past few, the clinical expertise has been successfully paired with the innovation of the Tucker Stem Cell Laboratory to advance the frontiers of science and medicine. These efforts first came to my attention at last year’s ARVO meeting in Seattle, where Budd Tucker and colleagues gave a number of impressive presentations that illuminated the power of this particular approach. The power lay not only in the ability to differentiate retinal cells from patient-donated skin cells, but to evaluate the genetic and molecular function within those cells and elucidate the disease manifestation in that specific patient. This combination of clinically informed, custom-made cell lines showed great promise for providing a model for testing targeted therapies, such as viral vectors carrying healthy copies of genes to take over the function that the patient’s mutated gene was failing to carry out. In the ensuing year, this research team has made great progress in testing virus-mediated gene therapies for several retinal diseases, even very rare ones.
So that’s great news with respect to the first challenge I mentioned. What about the gene size issue? The options for alleviating vision symptoms in USH2A (or USH2C, USH1D, or USH1F) are limited, but another new technology that’s emerged over the past year has given us new hope on this front.
This new method is a system of gene editing. Without getting too technical, it involves three components: a DNA-cutting enzyme, a targeting mechanism, and a short sequence of DNA to be inserted. When these three factors are introduced into a cell, the targeting sequence directs the enzyme to a precise location on the chromosome where cuts will be made. Once this happens, DNA repair mechanisms will kick in and attempt to rejoin the cut-ends. The short sequence to be inserted will be included in the newly repaired strand, and will remain in the DNA. The therapeutic impact here is that an identified genetic mutation can be cut out and replaced with ‘good’ code, much like you’d edit a single line of text using ‘cut’ and ‘paste’ features, or swap out a circuit on a motherboard. Here’s a simple figure to help visualize what’s going on:
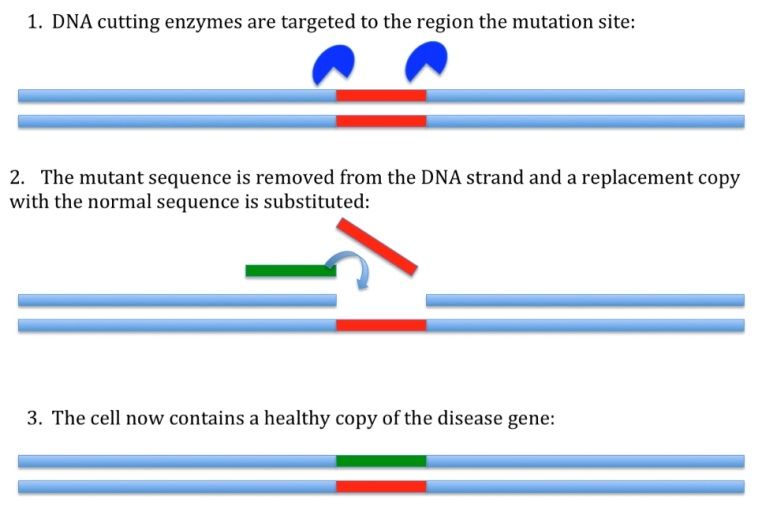
If you were doing this procedure in cultured retinal cells derived from an USH patient, you would be able to repair the USH mutation and generate a new line of cells that were identical to the patient DNA in every way except for that one tiny change. If those cells could then be transplanted to the patient, the likelihood of tissue rejection would be low (because the cell line originated from the patient’s own cells), and you would have effectively replaced the retinal cells lost to disease in that patient with new retinal cells bearing a functional copy of the affected gene.
The fact that cell culture models offer a really tractable chance at repairing and replacing defective genes that can’t be targeted with any other known method is exciting indeed. What’s more, Drs. Tucker and Stone believe that the cell culture model might provide an important step in being able to address the first challenge I mentioned, that of finding ways to help patients with diseases so rare that a commercial therapy probably isn’t in the cards. Remember that one of the limiting factors is the scarcity of patients to actually test the therapy on. The question usually addressed in Phase II/III clinical trials—does this repair (whether it be gene editing or viral delivery of a replacement gene or something else) actually do what it’s supposed to do in a patient with this particular genetic disease? -–is difficult to ask if you don’t have a statistically significant number of patients available with that particular genetic disorder.
But what if you could ask that question using stem cells derived from patients? What if, as part of your diagnosis, you gave permission for your biopsied skin cells to be converted into retinal stem cells and maintained in cell culture so that different therapies could be tried out and assessed on your particular type of mutation? You—or at least your genetically unique cell lines—could conceivably participate in multiple clinical trials simultaneously. You could literally be in 2 (or 4 or 16…) places at once, and one crucial limitation of developing treatments for rare diseases would be greatly reduced. Of course there would still have to be preclinical safety trials in animals, but with the stem cell strategy taking care of the gene-specific effectiveness question, those animals wouldn’t need to have the exact human mutation in order to show that the therapy was safe (i.e. not generally damaging to the targeted tissue or to any other part of the body). This research would go a long way toward removing some of the hurdles that limit the number of therapies that could even be considered for clinical trials, and that is exciting news indeed.
That’s it from this year’s ARVO, but happily the next opportunity to hear about the latest Usher research is just weeks away, at the International Symposium on Usher syndrome and Usher Family Conference. I look forward to meeting some of our blog readers there!







