
This section is a compilation of answers to the questions most commonly asked about Usher syndrome. Just start by following one of the links below for detailed answers. If you can’t find the question you wanted to ask, don’t hesitate to contact us at info@usher-syndrome.org.
- What is Usher syndrome?
- How many types of Usher syndrome are there?
- What is Usher Syndrome Type I?
- What is Usher Syndrome Type 2?
- What is Usher Syndrome Type 3?
- What is Retinitis Pigmentosa?
- What causes Usher syndrome?
- Does Usher syndrome always result in total blindness?
- Why is early diagnosis important?
- How does basic genetics apply to Usher syndrome?
- What is a genetic test?
- How are genetic tests performed?
- Why is genetic testing helpful?
- How does a child inherit a recessive mutation?
- How are the results of an Usher gene test interpreted?
- How much does a genetic test cost?
- What are the research areas into potential treatments?
- What hearing changes can occur?
- What is the impact of geography and cultural values
- Glossary of Terms
- Glossary of Common Terms - Clinical Trials
- Glossary - National Institute of General Medical Sciences
-
Usher syndrome is a genetic condition that causes hearing loss, vision loss, and sometimes balance/vestibular problems.
-
There are three clinical types of Usher syndrome.
-
Children with Usher type 1 are usually born profoundly deaf and experience progressive vision loss due to a retinal disease called retinitis pigmentosa (RP)...
-
Individuals with Usher type 2 are born hard-of-hearing and gradually lose their vision due to retinitis pigmentosa (also known as RP)...
-
Usher syndrome type 3 is is the rarest form of Usher syndrome, characterized by later onset hearing loss, RP that manifests between the second and fourth decades of life and variable vestibular dysfunction...
-
Retinitis pigmentosa (RP) is a disease of the retina cells in the eye, affect light sensitivity. RP can result in night blindness and tunnel vision.
-
Usher syndrome is inherited, which means that it is passed from parents to their children through genes. Usually, parents who have typical hearing and vision do not know if they are carriers of an Usher syndrome gene mutation.
-
Very few people with Usher syndrome will become totally blind – that is, have no light perception. Some adults maintain a small degree of central vision throughout their lives while others lose all usable vision.
Children will begin to develop night blindness in dimly lit areas. Progressive loss of visual fields will affect their ability to see to their right, left, above and below. -
In short, there is something that can be done. Lots of somethings. But you need a definitive diagnosis and you need it as early as possible.
-
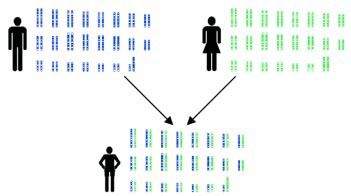
In total, humans have about 100,000 different genes that are grouped into small structures called chromosomes. People have 23 pairs of chromosomes, including a pair of sex chromosomes. Each pair consists of one chromosome that is inherited from the mother and another chromosome that is inherited from the father. The sex chromosomes contain genes that determine the sex of a person. Girls inherit two X chromosomes, whereas boys receive one X chromosome and one Y chromosome.
-
What is a genetic test?
A genetic test determines the DNA sequence of a certain region of the human genome. This region could be a whole gene, a portion of a gene, or other areas thought to regulate genes. The test will look for certain changes in the sequence that are known to have consequences on the function of a gene. These tests can be used to:
- diagnose a disease or other trait,
- determine if a person is a carrier of a mutation that could lead to disease in their children, and
- predict if a disease or trait that is not yet detectable by other medical tests may occur in the future.
-
Most genetic tests are performed on a DNA sample. Typically only 5-10 ml (1-2 teaspoons) are taken for a test. Some laboratories will allow cheek cells to be submitted for genetic testing. In this case, the cells are usually collected by rubbing the inside of the mouth with a small brush.
-
If you are visiting this website, chances are you suspect that you or a family member has Usher syndrome. The only way to know for sure is through genetic testing. Knowing can lead to improved decisions about treatment and management. In addition, genetic testing can help determine if other problems related to Usher syndrome besides hearing loss may be present or may develop in the future.
-
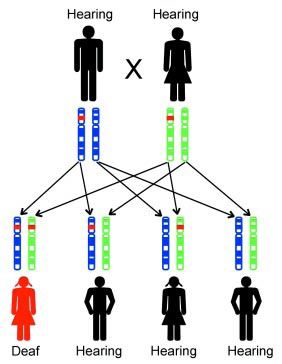
(chance of receiving mutation from father) x 1/2
(chance of receiving mutation from mother) x 1/2
= (chance of receiving two mutations) 1/4Therefore, in each pregnancy, there is a 25% chance that the child will inherit both mutations and, on average, 1 out of 4 children will be have Usher syndrome.
-
There are four possible outcomes to an Usher gene test: No mutations are detected, Two mutations are detected, Only one mutation is detected, or The mutation that was found may be unrelated to the hearing loss.
How are the results of an Usher gene test interpreted?
There are four possible outcomes to an Usher gene test:
No mutations are detected:
If no mutations are found, and the entire coding sequence was analyzed in a gene, it is unlikely that the hearing loss is caused by mutations in that specific gene. However, the patient may have Usher syndrome due to mutations in another gene that causes Usher syndrome. Not all genes for Usher syndrome have been identified.Two mutations are detected:
If two identical mutations or two different mutations in the same gene are found, and these mutations have been previously found to cause Usher syndrome, it can be assumed that the hearing loss is caused by these mutations.Only one mutation is detected:
If only one mutation is detected, interpretation can be difficult.- It is possible that the test did not detect the second mutation. Even though examining the whole coding sequence of the gene will detect most mutations, there are other regions of the gene sequence and surrounding DNA that could contain a mutation. Unfortunately, these sequences are rarely analyzed unless a specific mutation is already known.
- The mutation that was found may be unrelated to the hearing loss.
Mutations were detected but their significance is unknown:
Some changes in these genes are not considered to affect the function of the gene. These changes are often called “polymorphisms”. Sometimes, a new mutation is found and it is not yet clear whether the change will cause hearing loss or not. Unfortunately, more studies would need to be done before a definite conclusion could be made. -
How much does a genetic test cost?
The cost and turn-around-time of a genetic test may vary depending on the lab and the methods used for testing. Some Usher screens cost around $500. More accurate and detailed testing may cost between $2000-$5000. A typical range of time to get the results might be 8-12 weeks. Insurance companies will often pay for genetic tests, but you should check with your company before your doctor orders the test.
Individuals living in the United States who suspect they have Usher syndrome may be eligible for free genetic testing through the: Open Access Genetic Testing Program
When you receive your results, please be sure to update your USH Trust Registry.
-
Research into effective treatments for Usher syndrome is focused on four main areas: gene therapy, retinal implants, stem cell therapy, and drug-based therapy. For the latest news on these and other research, Science News.
For research updates organized by subtype, go to: Progress in Research to Treat Usher Syndrome
-
People with Usher syndrome often greatly benefit from hearing interventions such as hearing aids and cochlear implants. People with profound hearing loss often get little benefit from hearing aids. Those that choose cochlear implants have very little degradation in hearing as they age since cochlear implant bypass the hair cells in the cochlear.
-
Some genetic conditions occur more frequently among people whose ancestors are from specific geographic locations or religious/cultural groups. Due to either intermarriage or a small gene pool, there is a higher chance that specific genes common to this group will be passed on, including genes which cause specific conditions such as Usher syndrome. Because of this phenomena, Usher syndrome may be more frequently seen in certain ethnic groups such as Ashkenazi Jews, Acadian/French Ancestry, or Finnish heritage.
-
Glossary of Terms
Terminology related to Usher syndrome, blindness, hearing loss, and genetics is explained or defined below.
-
Learn about the words and terms used to describe clinical trials from the National Institutes of Health.
-
This glossary provides pronunciations and easy-to-understand definitions for terms commonly used in basic biomedical research. To search the glossary, enter the word or term you’re looking for and the terms and definitions containing the word(s) will appear below. Search results are refined with each letter entered. You can also use your browser’s “find” feature.
-
Audiologist
A person who specializes in testing hearing and hearing loss.
-
A person who has one typical, or unaltered version of a gene, and one version with a mutation. This person is not usually affected by the mutation, but is capable of passing it on.
-
Chromosome
A structure containing many genes arranged on a long strand of DNA. Each person has 23 pairs of chromosome, including a pair of sex chromosomes.
-
Clinical geneticist
A doctor who specializes in recognizing and treating patients with genetic diseases.
-
DNA
Deoxyribonucleic acid. The chemical that makes up genes. It is composed of adenine (A), cytosine (C), guanine (G), and thymidine (T).
-
Gene
A unique sequence of DNA that serves as a specific set of instructions in the body.
-
Hearing threshold
The lowest level of sound that can be heard during a hearing test.
-
Hereditary
Inherited; something that is passed from parent to child through the parent's genes
-
Mutation
A change in a gene sequence that often disrupts the function of the gene.
-
Mutation - Recessive
A mutation in a gene that is not strong enough to make a person affected if the person also has a typical copy of the gene.
-
Nonsyndromic Usher syndrome
This diagnosis occurs when a person's genetic test reveals that they have two of the same Usher genes, but they do not exhibit all of the symptoms associated with that type of Usher syndrome. For example, a person may have two USH2A genes, but only have retinitis pigmentosa (RP) with no hearing loss. OR, a person may have two USH1C genes, but only experience profound deafness with no RP, and no balance issues. A syndrome means two or more symptoms always occur together. If a person only has one symptom, it is no longer a "syndrome."
-
Ophthalmologist
A doctor who specializes in studying eye diseases.
-
Otolaryngologist (ENT/ORL)
A doctor who studies ear, nose and throat disorders.
-
Sensorineural hearing loss (SNHL)
Hearing loss caused by damage to the inner ear or auditory nerve
-
Syndrome
A set of symptoms or conditions that occur together and suggest the presence of a certain disease. The symptoms or conditions related to Usher syndrome are hearing loss, vision loss caused by RP, and, sometimes, vestibular or balance issues.







