
Usher syndrome, Part II: A Complex Molecular Picture
August 12, 2011
By Jennifer Phillips, Ph.D.
In Part I I gave a rough overview of Usher syndrome and went over a bit of cell physiology of auditory and visual sensory cells. In this post, I'll introduce the molecules known to be affected in Usher patients, and begin to describe what is known about their function.
As mentioned previously, variations in the clinical presentation of Usher syndrome have resulted in the creation of three clinical subtypes. Type 1 is characterized by severe to profound congenital hearing loss, balance problems, and early onset vision loss, usually beginning before the patient's 10th birthday. The type 2 hearing loss is also congenital, but tends to be less severe. Although the vision loss in type 2 can become as severe over time as that seen in Type 1 patients, the first symptoms usually begin to occur a bit later, in the early teen years. Finally, these patients do not present with balance problems. In Type 3 patients, all three categories of symptoms-vision, hearing, and balance, are progressive, with even the hearing loss commencing in childhood or adolescence rather than at birth.
Despite these differences, the specific combination of deaf-blindness, plus or minus balance defects, led researchers to predict that multiple mutations in a single gene, or perhaps in two or three genes at the most, would turn out to be responsible for the symptoms observed in all Usher patients. This is the case, for example, with Cystic Fibrosis. Just like Usher syndrome, CF is a recessive disease, meaning that you need two defective copies of the gene to show the disease symptoms. Importantly, even though there is a remarkable degree of variation in symptom severity, the vast majority of CF cases are due to mutations in only one (albeit extremely large) gene.
With respect to Usher syndrome, the advent of genetic mapping led to surprising results in pinpointing the genetic underpinnings of the disease. To date, at least 11 different genes have been implicated in USH, nine of which have been molecularly identified thus far. More surprising still is the wide variety of proteins encoded by the identified genes. In contrast to standard signaling or metabolic pathways, in which a genetic defect at any point in a linear chain of events will result in the same basic problem downstream, the Usher proteins do not appear to act in stepwise fashion to regulate a cellular process. What they actually do, in fact is not entirely clear, but the various proteins contain functional domains known to be important for:
scaffolding: providing a structure on which multiple proteins can convene to assemble into a complex.cell adhesion: spanning the minute distances between a cell and its neighbor.signaling: molecular chitchat between cells in the same neighborhood.extracellular matrix components: proteins that confer structural support to the regions around cells.motor activity: the job of transferring molecules from where they're made to where they're going to work in the cell.membrane integration: a protein that inserts itself into the membrane of a cell, sometimes weaving in and out multiple times. Transmembrane proteins perform a wide variety of functions, so this characteristic alone tell us only where to find it, not what it's doing.
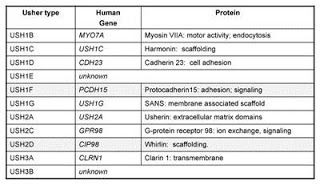
Table of the known Usher genes and protein identities.
Structural information about the Usher proteins provide a starting point from which researchers can begin to investigate their localization within the various working parts of the sensory cells affected in Usher syndrome, their potential physical interactions with one another and, ultimately, the role of these proteins in sensory cell function and survival.
Antibodies to these proteins enable us to visualize their exact position within the cell, and such experiments show that most of the Usher proteins colocalize to the very structures that are involved in specific sensory cell function-the photoreceptor connecting cilium , the stereocilia of the mechanosensory hair cells, and the specialized, neurotransmitter-laden synapses of both cell types. Laboratory experiments with the various functional domains of these proteins have further shown that they have the ability to physically interact and bind to one another to form a protein complex.
So, what might this motley crew of proteins be doing hanging out together at these very specific subcellular locations? The function of the Usher complex at the stereocilia has been fairly well studied using mouse or rat models of the disease to examine the effects of depleting the function of any one Usher gene. In the first panel of the figure below, hair cells from the cochlea of a young mouse with no defective Usher genes are shown to have rows of stereocilia that are nicely arranged in a crescent shape, as well as consistent length, width, and orientation of the individual stereocilia. In contrast, the stereocilia of mice with mutated forms of Usher genes (all Type 1 genes in this case) are dramatically misshapen and disorganized. ???
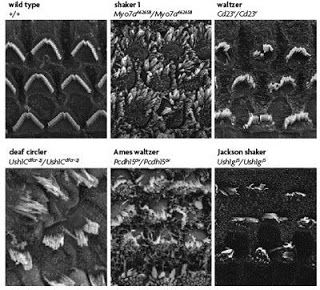
from Brown et al, 2008. The top left panel shows an electron micrograph of a normal, healthy mouse cochlea. Above each additional panel, showing cochleae from Usher mice, is the name of the mutant line, chosen based on the observed behavior. Because of the balance problems, these mice tend to run in circles and toss their heads. The affected gene in each case is listed below the name.
?The subcellular localization of Usher proteins in hair cells, illustrated in the schematic drawing below, provide insights to how such disorganization might come about. Usher protein localization in the stereocilia are shown at three timepoints ranging from late embryonic to several weeks postnatal.
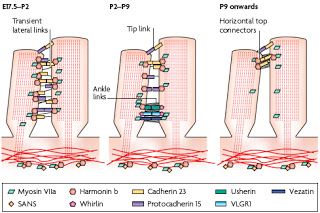
From Brown et al, 2008. This is a cartoon of highly magnified stereocilia, just showing the relationship between two neighboring stereocilia projecting from the same hair cell. Each little colored shape represents an Usher protein or a known interacting protein. Three different developmental time points are shown, from left to right. The stereocilia on the far left represents a time spanning from late gestation to newborn, followed by representations of two additional postnatal time periods. Note that the location of and relationships between the little colored shapes changes quite a bit over the course of this two week period.
It's important to note here that baby mice are born deaf and do not begin to hear until about the 6th postnatal day. The dynamic localization of the Usher proteins in the schematic above, in which many of them appear to move from one part of the stereocilia to another over time, serves to illustrate that even after birth there is still quite a bit of fine-tuning going on both structurally and functionally with respect to the stereocilia in the mouse. Human auditory development occurs on a different time scale (not surprising considering that mouse gestation only takes 21 days versus 40 weeks in humans); fetuses can hear sounds in utero for several months prior to birth, but it's likely that a similar process occurs in the growth and maturation of stereocilia in human hair cells prior to the 30th week of gestation.
Thus, noting that the presence of Usher proteins is required for normal growth and patterning of the stereocilia prior to the acquisition of hearing, it is easy to understand why the hearing loss in patients with mutated copies of these Usher genes is congenital. These data also indicate that although Usher proteins may also play a role at the hair cell synapses, based on their expression there, the primary cause of deafness, at least in Usher type 1 patients, is probably due to the perturbations of the stereocilia. These changes in stereocilia structure and function can also explain the balance defects associated with the type 1 and type 3 forms of the disease, but it still isn't clear why patients with mutations in any of the three Usher type 2 genes dodge this particular symptom.
Compared to the situation in the ear, the functional importance of Usher proteins in the retina is relatively poorly understood, but I'll summarize the findings thus far in Part III, and then wrap up with some information about clinical diagnosis and treatment options in part IV.







